Conclusions

Presentation Drafts
First Draft
|
Second Draft
|
Third Draft
| ||||||
What is Griscelli Syndrome Type II?
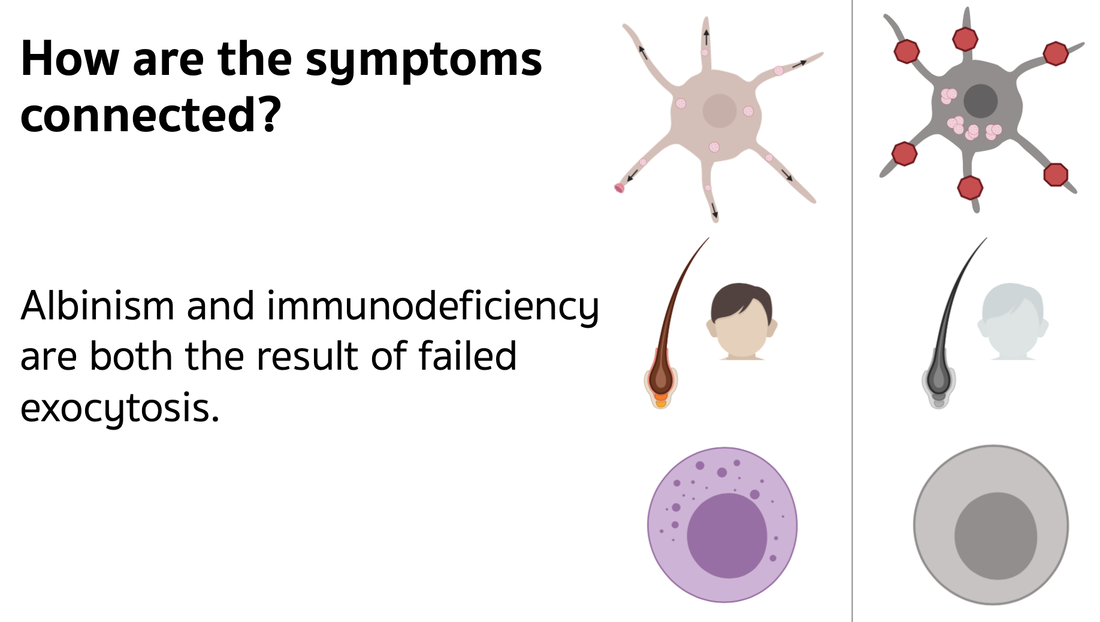
|
Griscelli Syndrome (GS) Type II is a genetic disease with an autosomal recessive inheritance pattern. The key symptoms are partial albinism and immunodeficiency. The partial albinism gives patients silver hair and loss of pigmentation. The immunodeficiency is not the result of decreased immune cell count but rather defective immune cells of normal counts.
|
What is the cause?
|
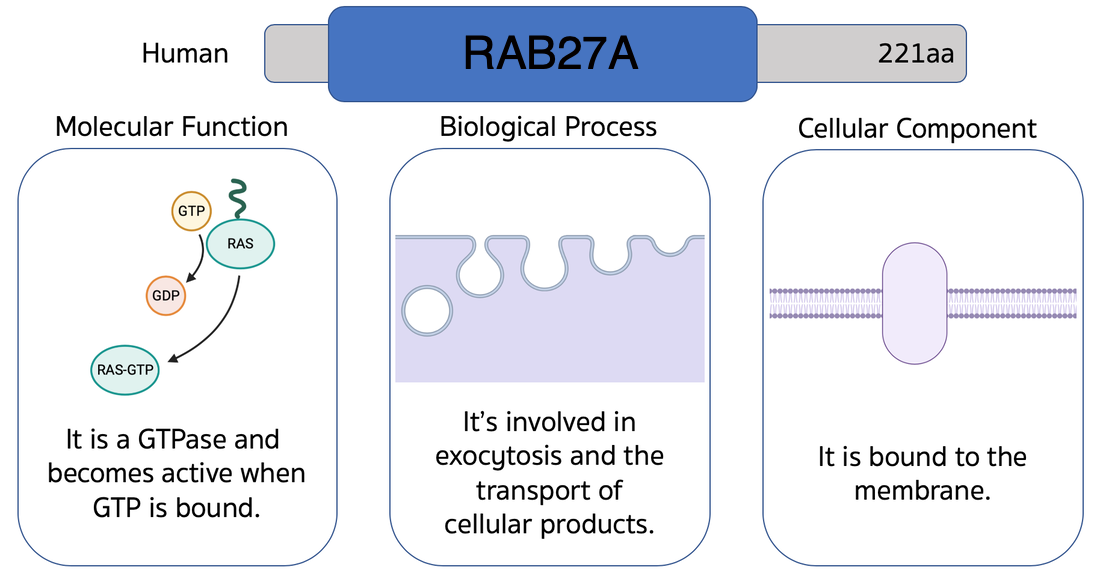
Type II is the result of mutations in the gene RAB27A. A gene that encodes a membrane bound GTPase that is understood to play a role in the pathway of exocytosis in cells.
|
|
|
|
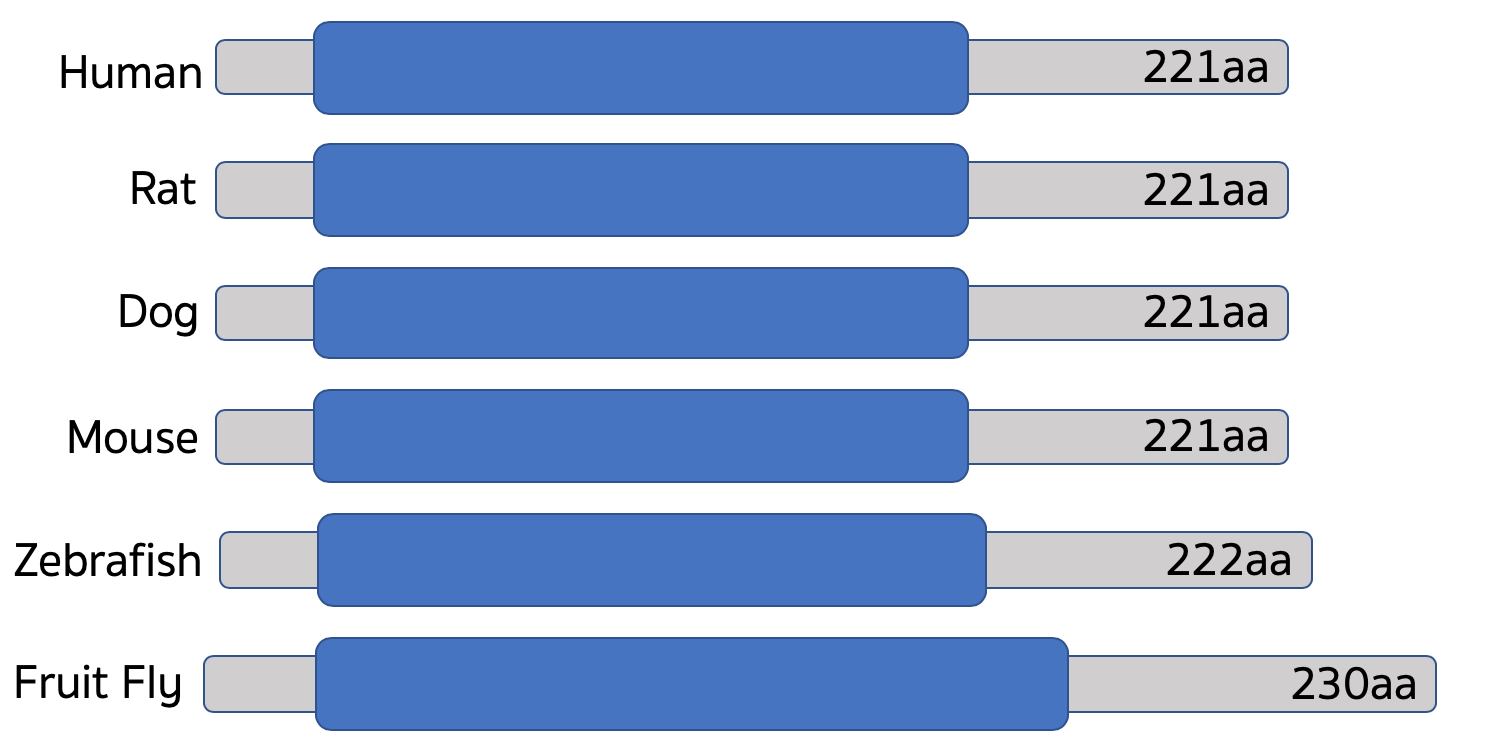
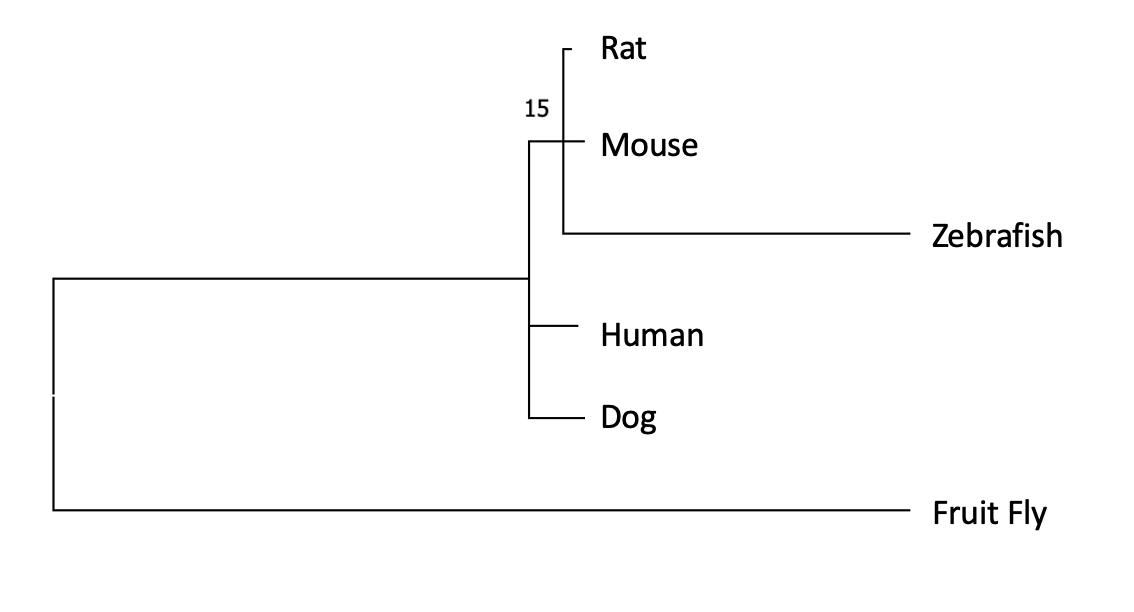
Homologs of RAB27A can be found in many species cutting across many groups of organisms which indicates the gene likely evolved some time ago and has been retained in descendants to this day.
The architecture of the gene is extremely simple and well conserved between species which gives confidence that it functions very similarly between species and that results taken from studying model organisms will be applicable to humans.
The architecture of the gene is extremely simple and well conserved between species which gives confidence that it functions very similarly between species and that results taken from studying model organisms will be applicable to humans.
Mice as a model organism for studying GS
|
|
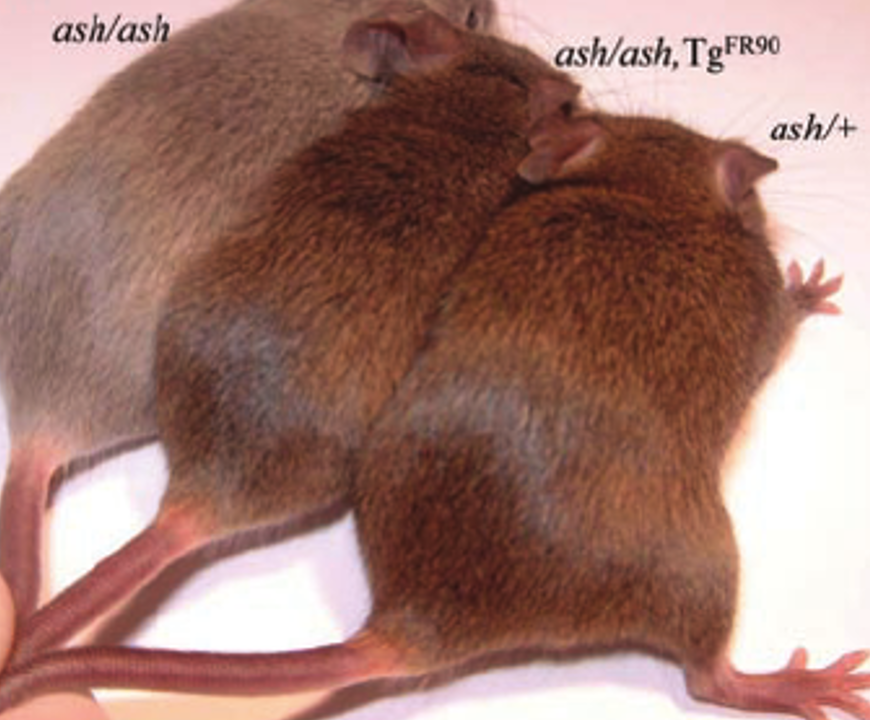
Mice make an effective model organism for studying GS in part because previous work has established that mice with mutated RAB27A, referred to as ashen, show a silvery coat closely paralleling the partial albinism that is a symptom of GS. This easy to spot phenotype makes identification of mutants easy in experiments and it gives a starting to point to build off of rather than having to start over with a new model organism.
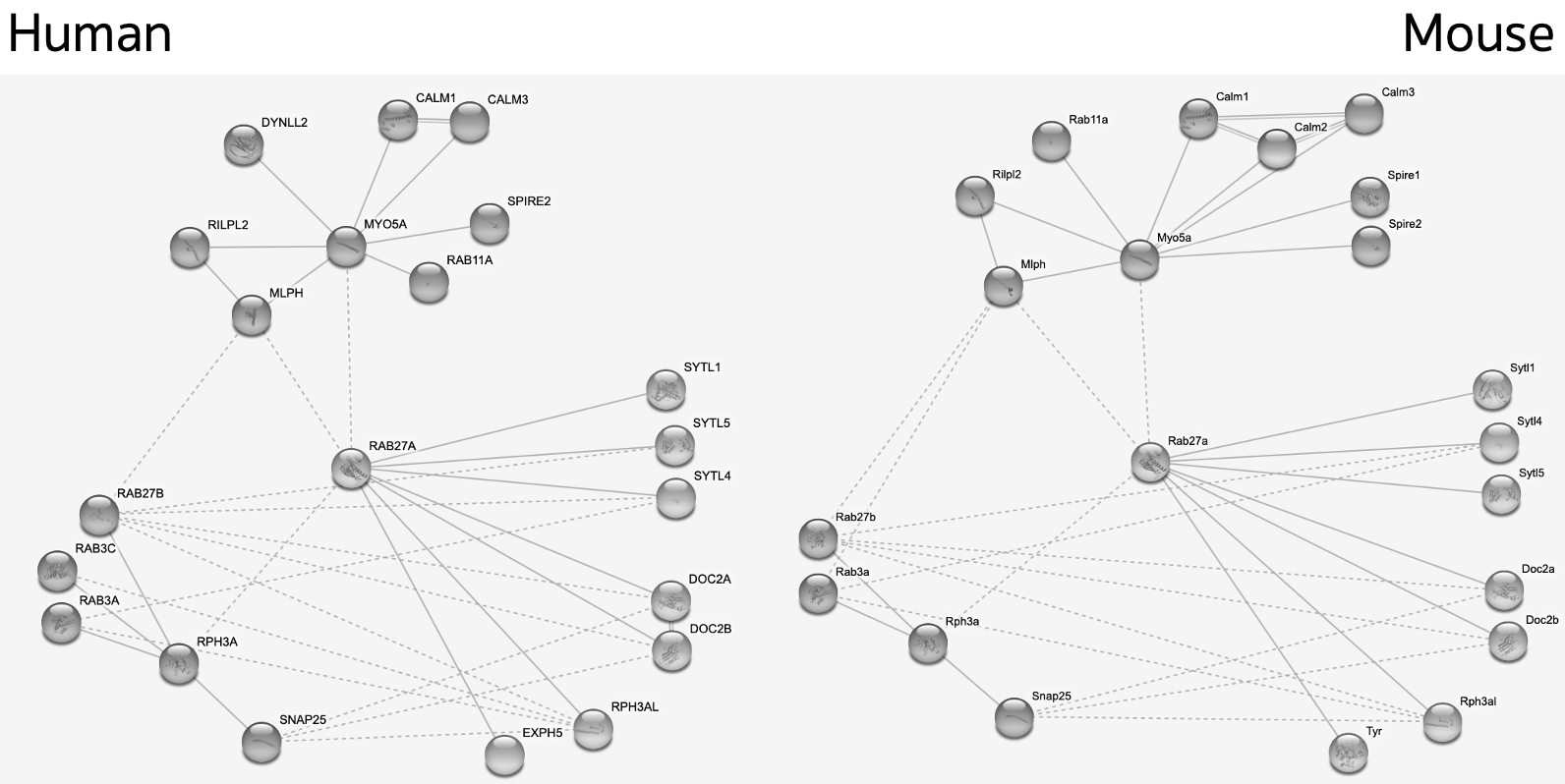
Additionally, human and mouse copies of the protein have been shown to have incredibly similar protein interaction networks which allows mice to be an effective proteomic proxy for humans.
Additionally, human and mouse copies of the protein have been shown to have incredibly similar protein interaction networks which allows mice to be an effective proteomic proxy for humans.

Addressing the gap in knowledge
|
|
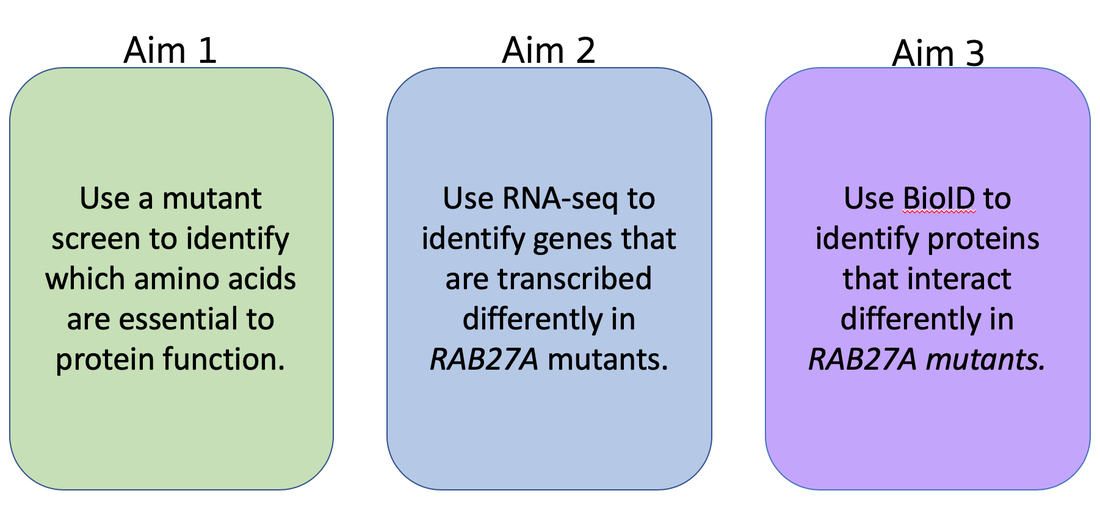
Aim 1: Identify essential amino acid differences in individuals with loss of RAB27A function
|
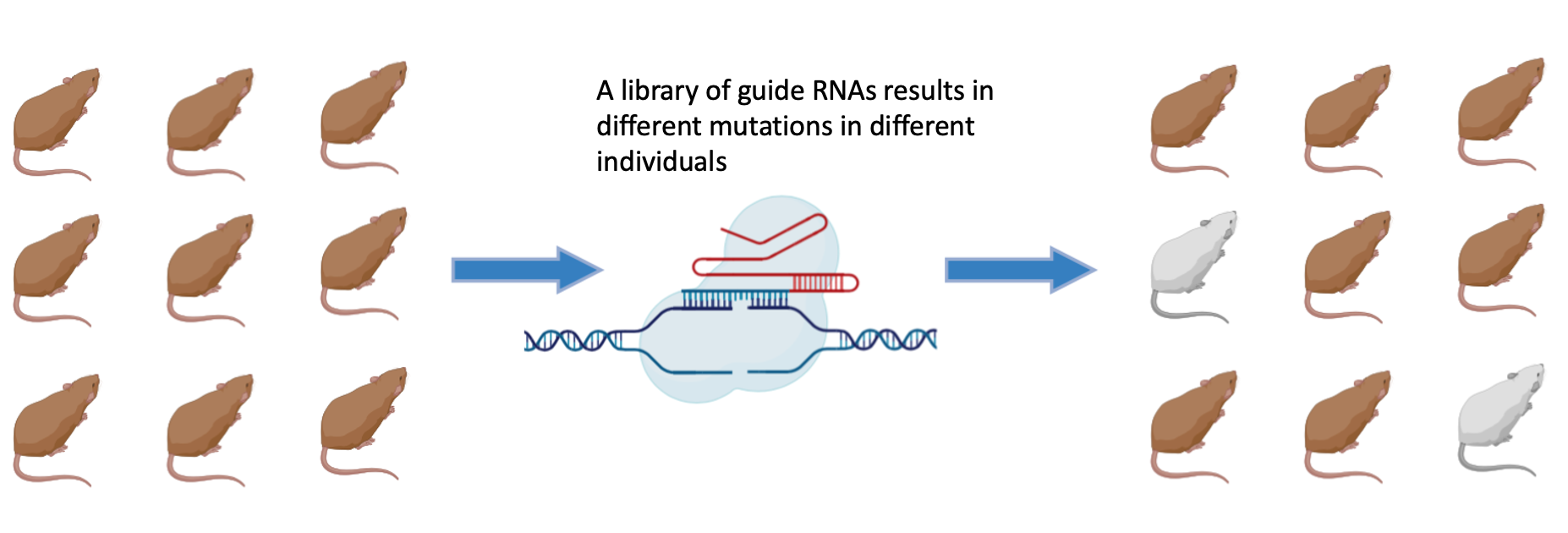
Using a CRISPR/CAS9 system wild type (WT) mice will be mutagenized with the goal of knocking out RAB27A function with various mutations for the purposes of a genetic screen. Successfully mutated mice will be identified by their silvery coat.
|
|
Successfully identified mutants will then have their genome sequenced to determine the nature of the mutation that knocked out RAB27A function and which amino acids it affected. This procedure seeks to probe the architecture of the gene and attempt to discover more about the reactivity of the protein. This is made more difficult by the fact that the gene doesn't have multiple domains to manipulate in search of domain specific effects on the phenotype. Instead I seek to use the specific amino acids that are found to be essential for the genes function and use their known patterns of reactivity to make inferences about the reactivity of the assembled protein.
Hypothesis: Single nucleotide polymorphisms in key parts of the sequence of RAB27A will lead to loss of function mutations and the known reactivity of those amino acids will allow the generation of further hypothesis of which chemical reactions in the cell are part of wild type RAB27A function and if tissue specific chemical conditions might play a role in the tissue specific symptoms of GS.
Hypothesis: Single nucleotide polymorphisms in key parts of the sequence of RAB27A will lead to loss of function mutations and the known reactivity of those amino acids will allow the generation of further hypothesis of which chemical reactions in the cell are part of wild type RAB27A function and if tissue specific chemical conditions might play a role in the tissue specific symptoms of GS.
|
|
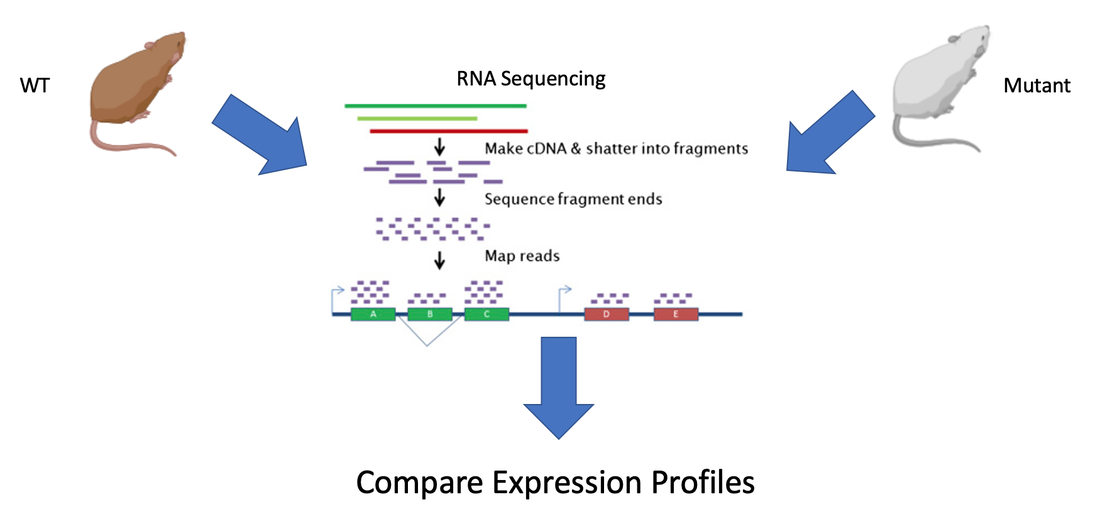
Aim 2: Identify differently transcribed genes that are implicated in exocytosis failure
|
RNA sequencing will be used to compare the RNA profiles of ashen mutant mice to wild type mice. Differences in the expression profiles will identify genes that are differently transcribed in mutants.
|
|
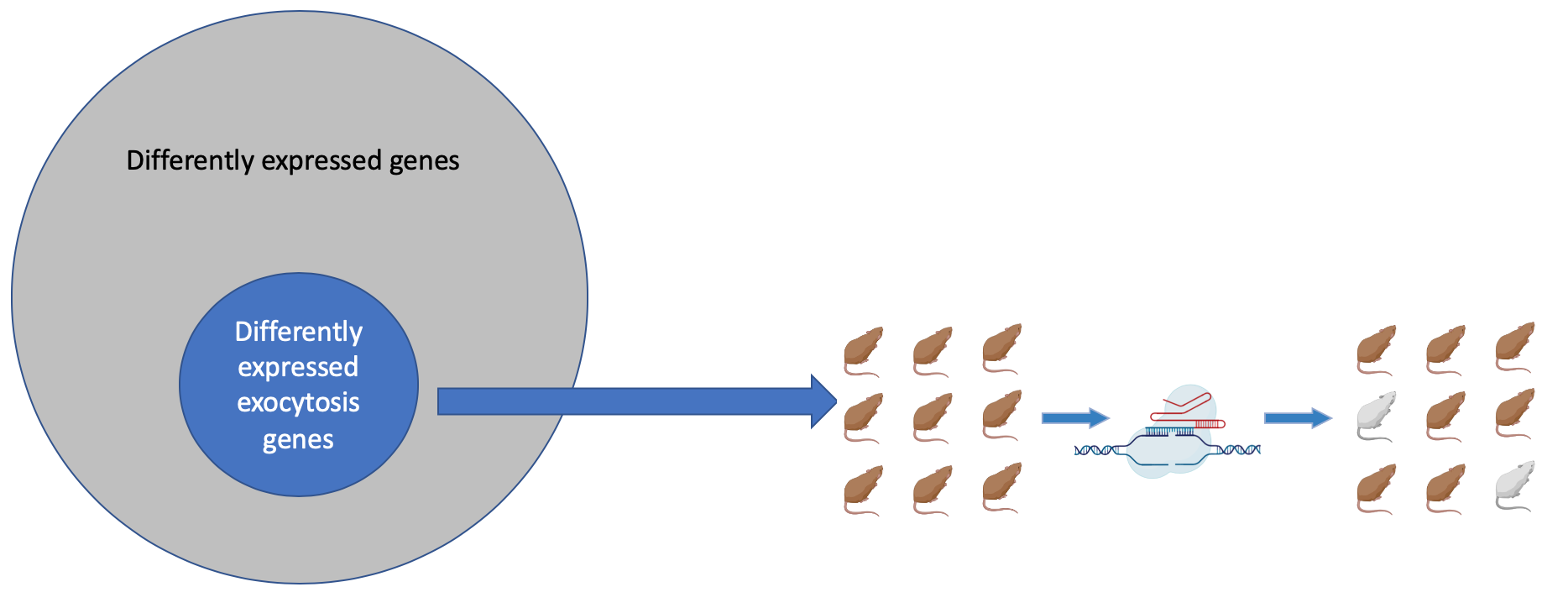
Once the differently transcribed genes are identified through RNA sequencing they will be sorted using Gene Ontology (GO). GO allows us to identify the subset of the differently transcribed genes that are involved in exocytosis.
|

|
|
Differently transcribed genes that GO predicted to have a role in exocytosis will then be knocked out in WT mice using CRISPR. The mutated mice will be visually assayed for a silvery coat. The presence of a silvery coat would confirm that those genes whose expression is RAB27A dependent play a role in exocytosis and without them melanin is not transported outside the cell just like in GS.
|
Hypothesis: Genes differentially expressed in ashen mutants will show tissue specific roles or expression involving hair and the immune system which may play a role in the tissue specificity of GS symptoms.
|
|
Aim 3: Identify differences in proteins interacting with RAB27A as part of the exocytosis pathway
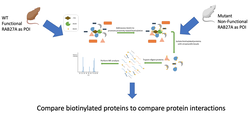
By using BioID and coupling biotinylation to RAB27A in both mutant and WT mice we can establish a protein interaction profile for both mutant and WT mice and compare them to look for differently interacting proteins. These differently interacting proteins might themselves have tissue specific patterns of expression that help explain GS symptoms.
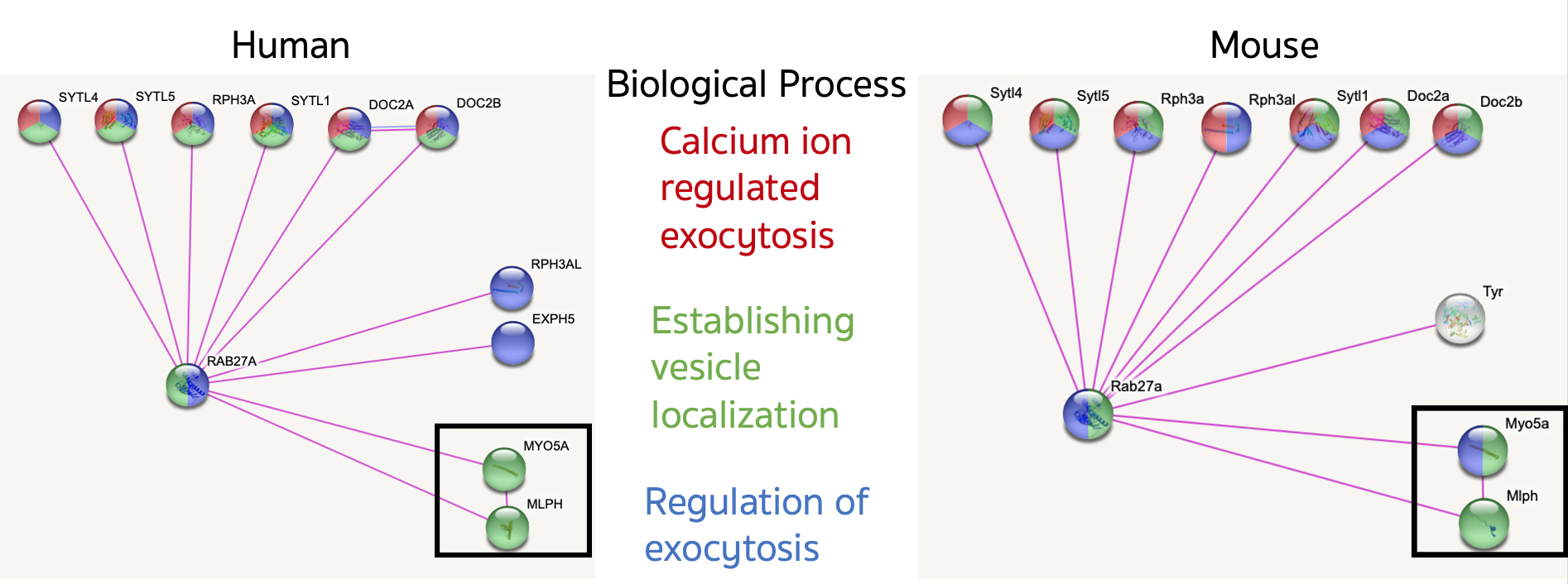
Of particular interest in the established WT protein interaction network is the complex RAB27A forms with MYO5A and MLPH in both humans and mice. If in the mutant mice that complex fails to form it might offer further explanation as to the exact reason for exocytosis failure in mutants. If the mutants interact with new proteins in their interaction network it could also serve to explain why exocytosis fails.
Hypothesis: Differentially interacting proteins in mutant RAB27A networks will show tissue specific roles or expression involving hair and the immune system which may play a role in the tissue specificity of GS symptoms.
Hypothesis: Differentially interacting proteins in mutant RAB27A networks will show tissue specific roles or expression involving hair and the immune system which may play a role in the tissue specificity of GS symptoms.
|
|

Future Directions
Given that the current best treatment involves bone marrow transplant I think it would be interesting to explore what happens in patients before and after transplant. Does the presence of transplanted marrow cells change the activity and behavior of neighboring native cells?
How can the methods and findings of research on this extremely rare disease help accelerate research on other extremely rare diseases and how can medicine be personalized to help people with conditions that don't often get much attention or funding?
How can the methods and findings of research on this extremely rare disease help accelerate research on other extremely rare diseases and how can medicine be personalized to help people with conditions that don't often get much attention or funding?

The web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison
